1. Introduction
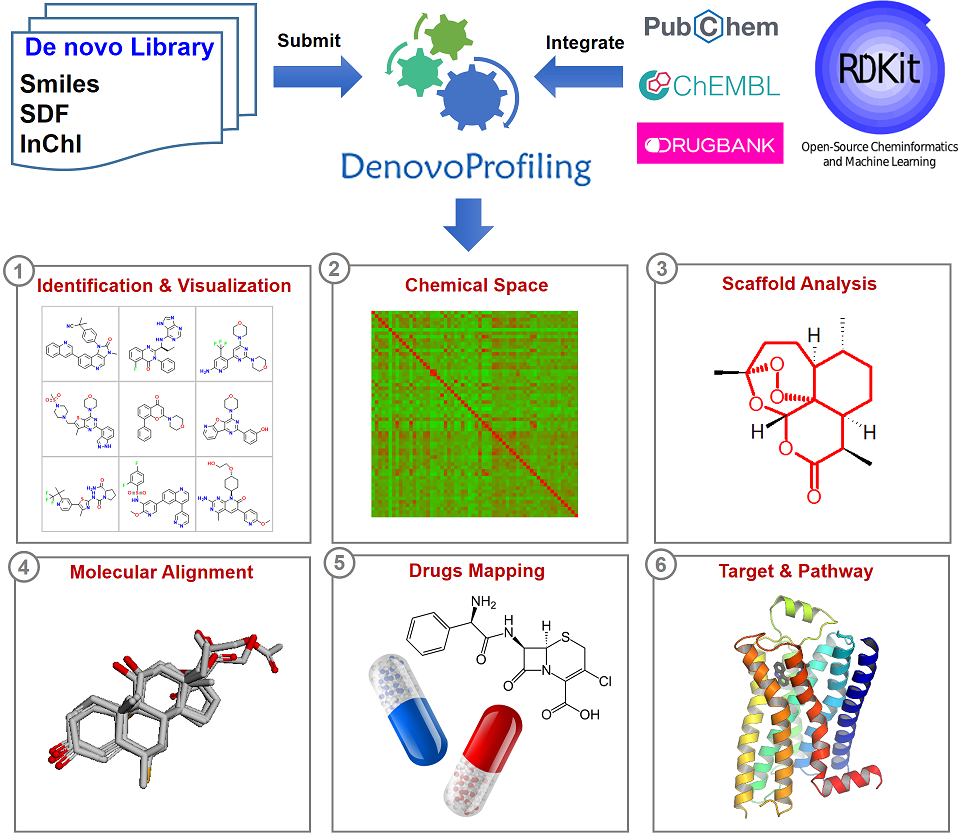
DenovoProfiling, a webserver dedicated for de novo generated molecule library profiling. We aim to provide a user-friendly public webserver to support the structure and chemical space visualization, scaffold analysis, molecular alignment, drugs profiling, targets and pathway profiling. We integrated cheminformatics tools and databases to provide comprehensive annotations for the de novo generated molecules. We believe that DenovoProfiling could be an efficient tool for user to capture the knowledge of de novo generated molecules quickly.
2. Framework of DenovoProfiling
3. Supported Chemical Formats
Four widely used chemical formats are supported in DenovoProfiling: SDF, SMILES, InChI, and CDX. All those formats files can be uploaded or the file contents can be pasted and submitted to the web server, except for the binary CDX format, which cannot be pasted. The Open Babel (www.openbabel.org) program was used for chemical file format conversion.
| Formats | Description | Upload | Paste |
|---|---|---|---|
| SDF | A chemical-data file formats developed by MDL, for holding information about the atoms, bonds, connectivity and coordinates of multiple molecules. | ||
| SMILES | The simplified molecular-input line-entry system (SMILES) is a specification in form of a line notation for describing the structure of chemical species using short ASCII strings. | ||
| InChI | The IUPAC International Chemical Identifier (InChI) is a textual identifier for chemical substances, designed to provide a standard and human-readable way to encode molecular information. | ||
| CDX | A binary file type created by CambridgeSoft Corporation's ChemDraw chemical structure application. |
4. Modules
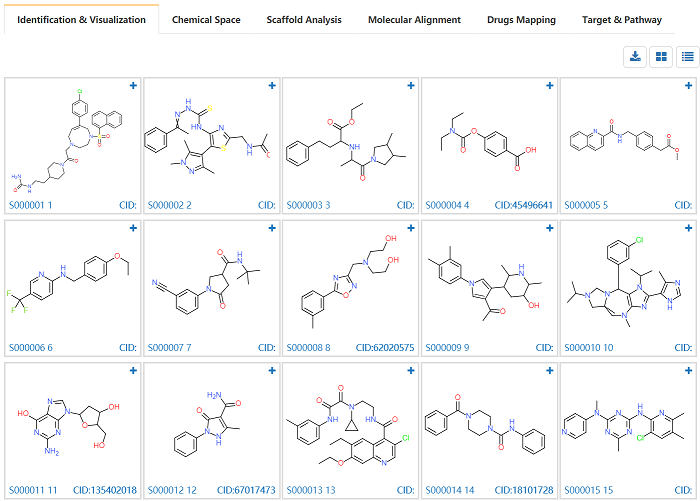
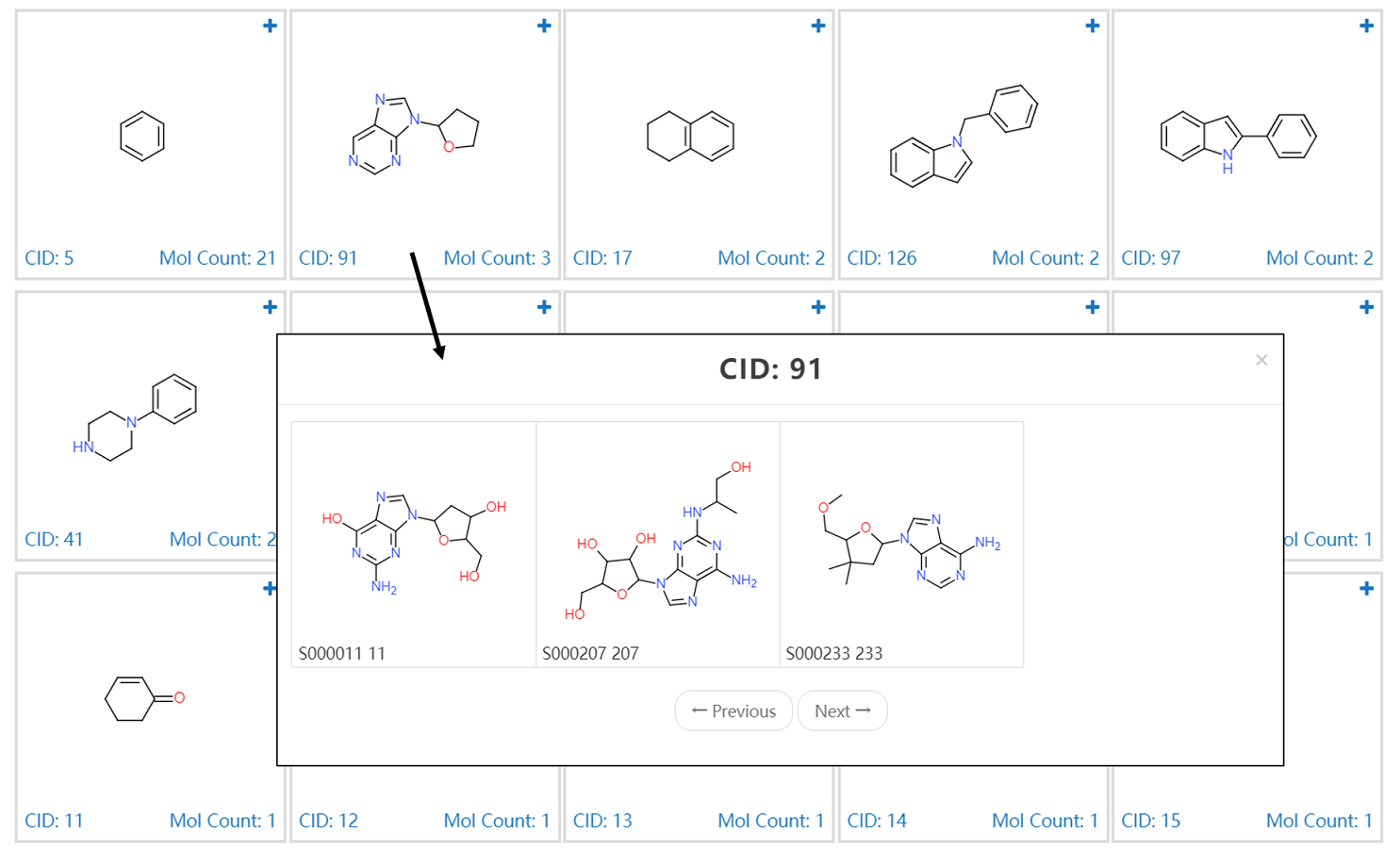
4.1 Identification & Visualization
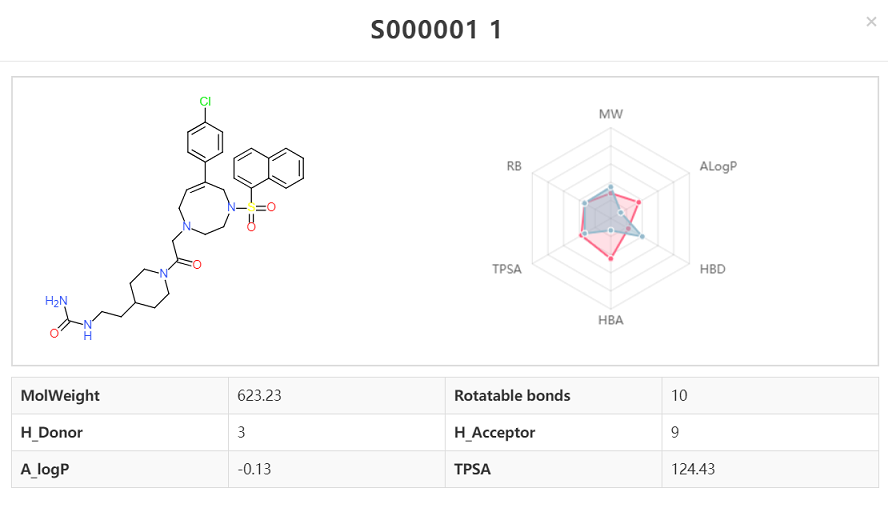
User could browse the structures with mapped PubChem Compound ID (CID). The properties including molecular weight, LogP, HBA, HBD, number of rotatable bonds, TPSA are given by clicking upright plus button. The CID is provided at bottom right and linked the PubChem which provides more detailed compound information.
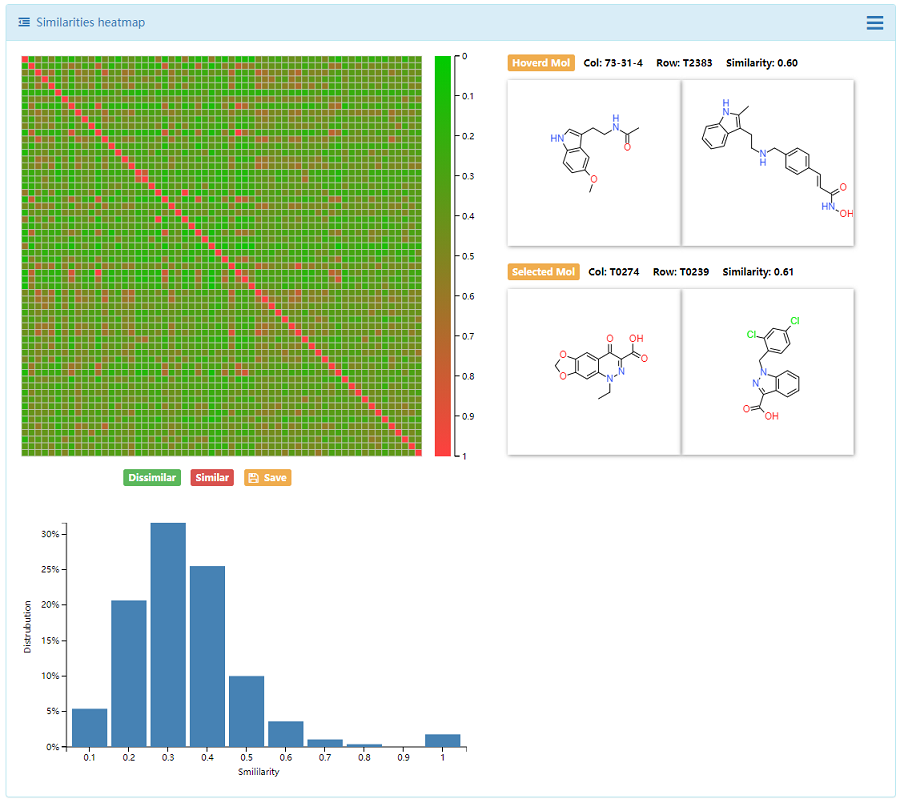
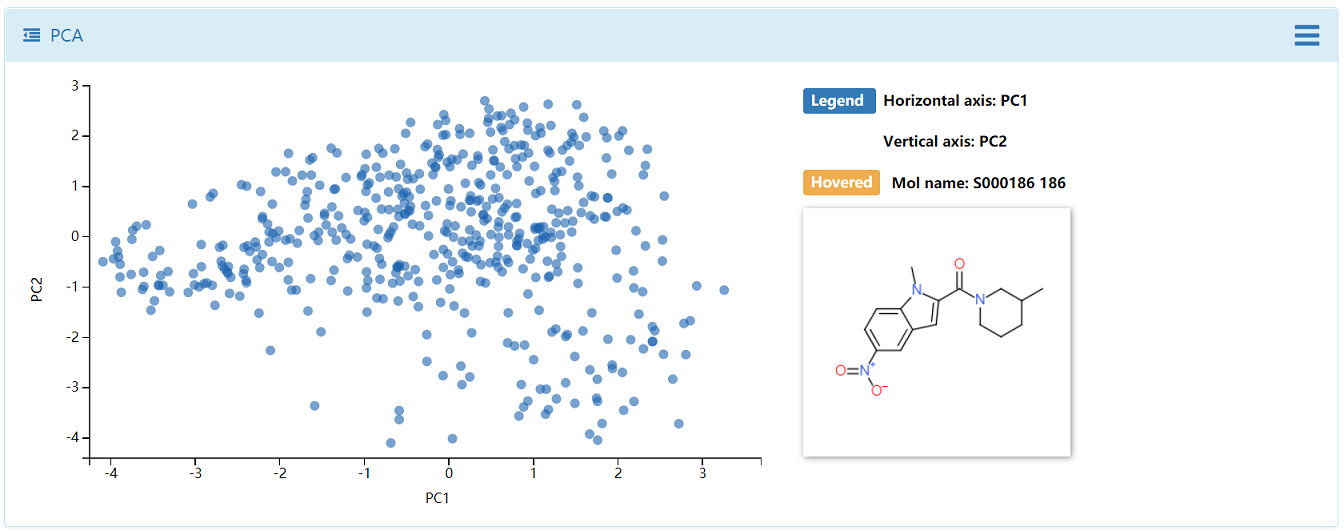
4.2 Chemical Space
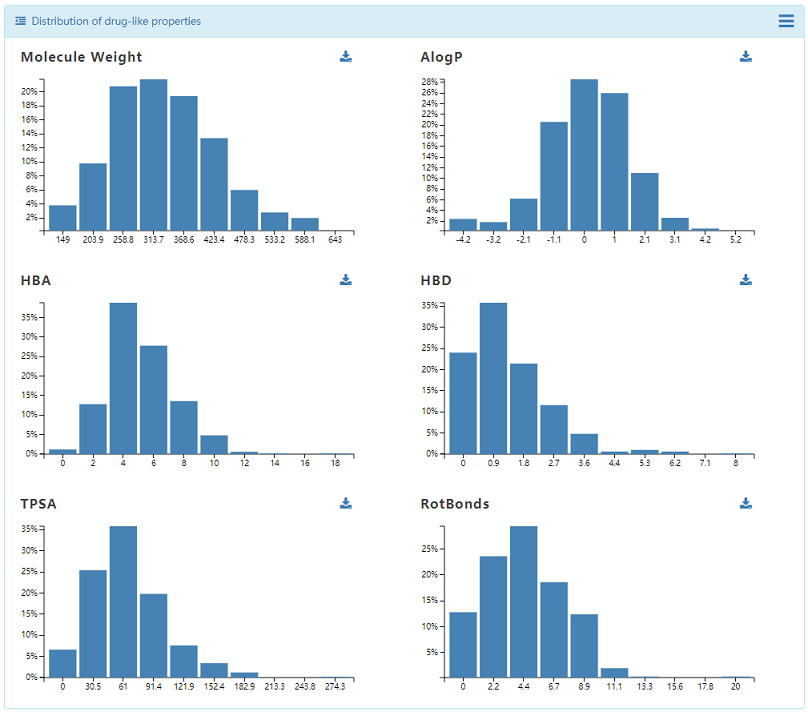
Two important approaches:similarity maps and principal components analysis (PCA) were used in DenovoProfiling. The generated similarity heatmap and PCA plots are interactive, which user could move mouse to the target of point, and the corresponding structure is visualized. Meanwhile, the distribution of drug-like properties was also plotted.
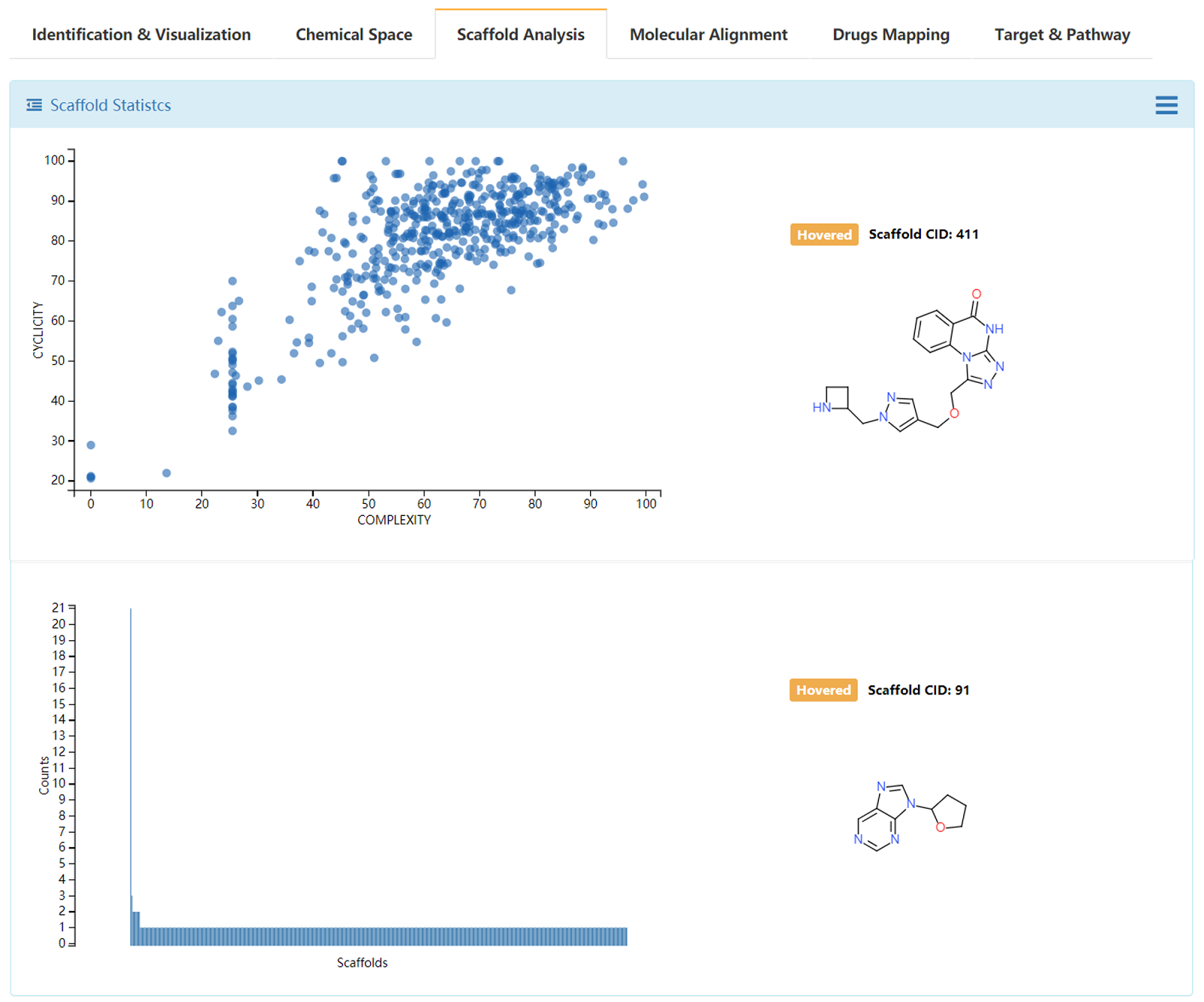
Bemis and Murcko (BM) scaffold approach was used to generate the scaffold for de novo generated library. The complexity and cyclicity of the scaffolds and statistics of each scaffold were interactive illustrated with scatter plot and histogram plot. The structures of scaffolds and their number of molecules were illustrated in grid table. The members of molecules for each scaffold could be browsed by clicking the upright plus button.
4.3 ADMET Prediction
Early estimation of ADMET(Absorption, Distribution, Metabolism, Excretion, and Toxicity) in the discovery phase could reduce the fraction of pharmacokinetics-related and toxicity-related failure in the clinical phases. we collected 13 ADMET datasets (Caco2 Cell Permeability, P-gp Inhibitors, P-gp Substrates, Biodegradability, CYP1A2, CYP3A4, CYP2D6, CYP2C9, CYP2C19, Liver Toxicity, HERG, Acute Oral Toxicity, Carcinogenic Potency) and constructed deep learning models using message passing neural network (MPNN) for ADMET profiling of the de novo generated molecules.

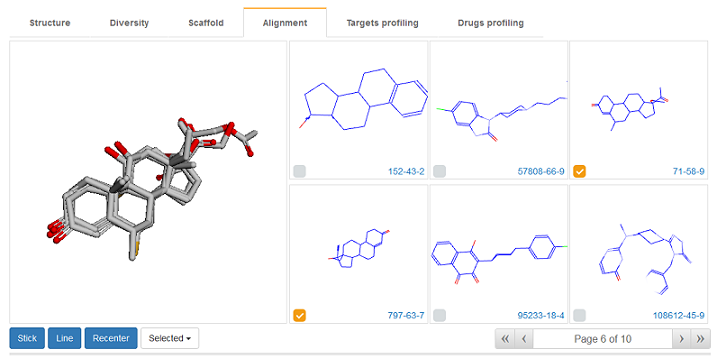
4.4 Molecular Alignment
Shape and pharmacophore combined approach in WEGA was used in DenovoProfiling for molecular alignment. User can upload a library of 3D structures, and DenovoProfiling would align all the structures to the first structure of the library. User could browse the alignment results, and select the molecules of interest to see the alignment result.
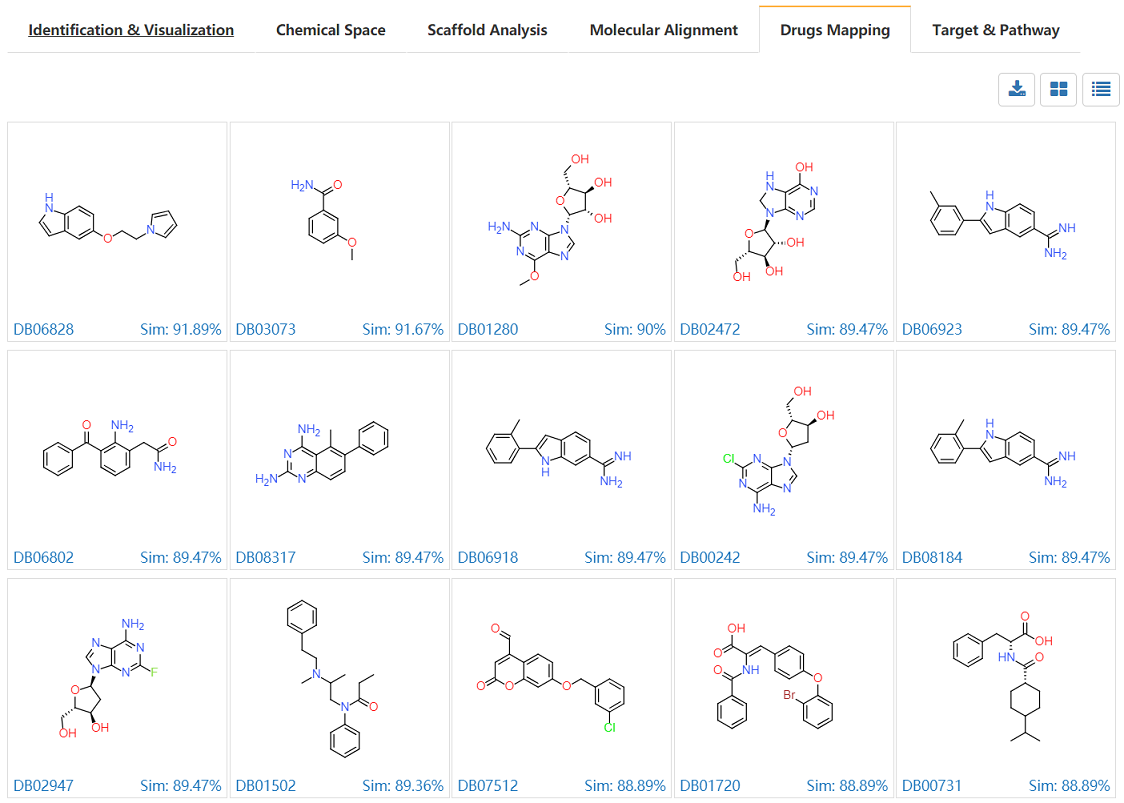
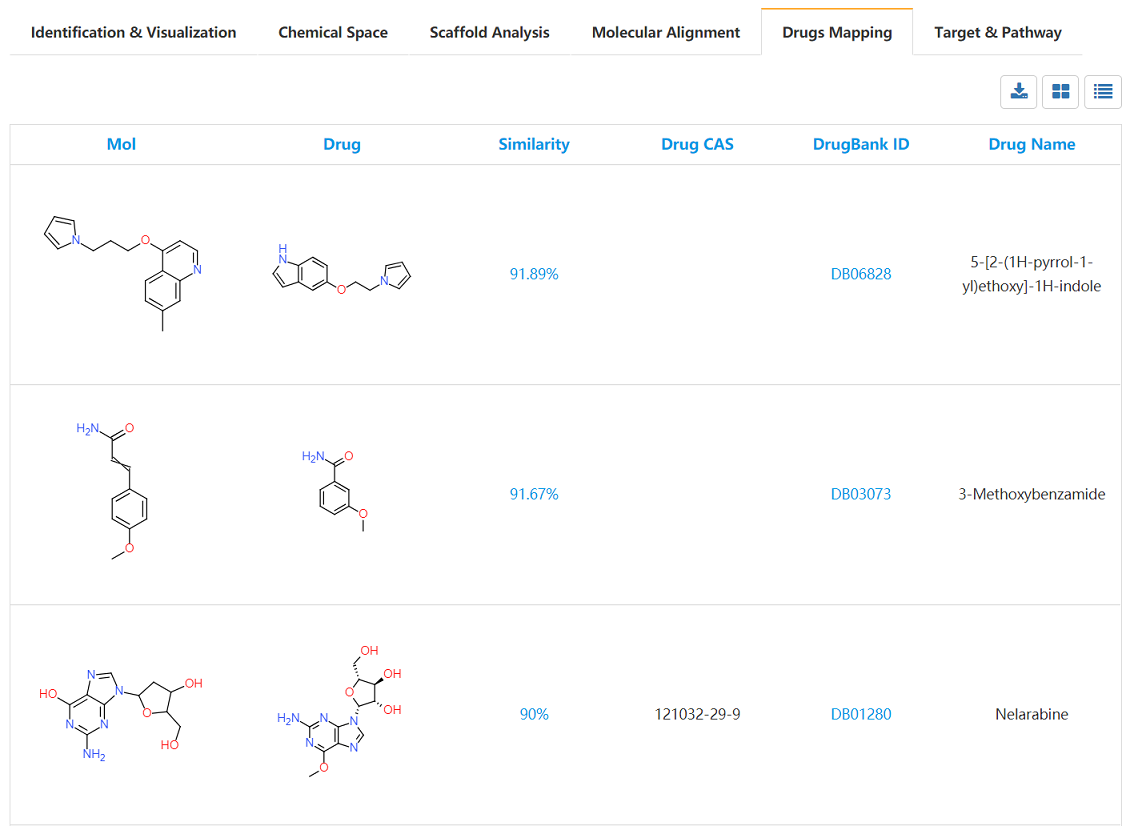
4.5 Drugs Mapping
Drugs structures library were prepared from DrugBank database, and the similarity calculations were carried on between the submitted de novo library and drug library. The grid view and the table view are provided. The DrugBank ID are also provided and linked to original database. Details of these drugs information could be obtained directly.
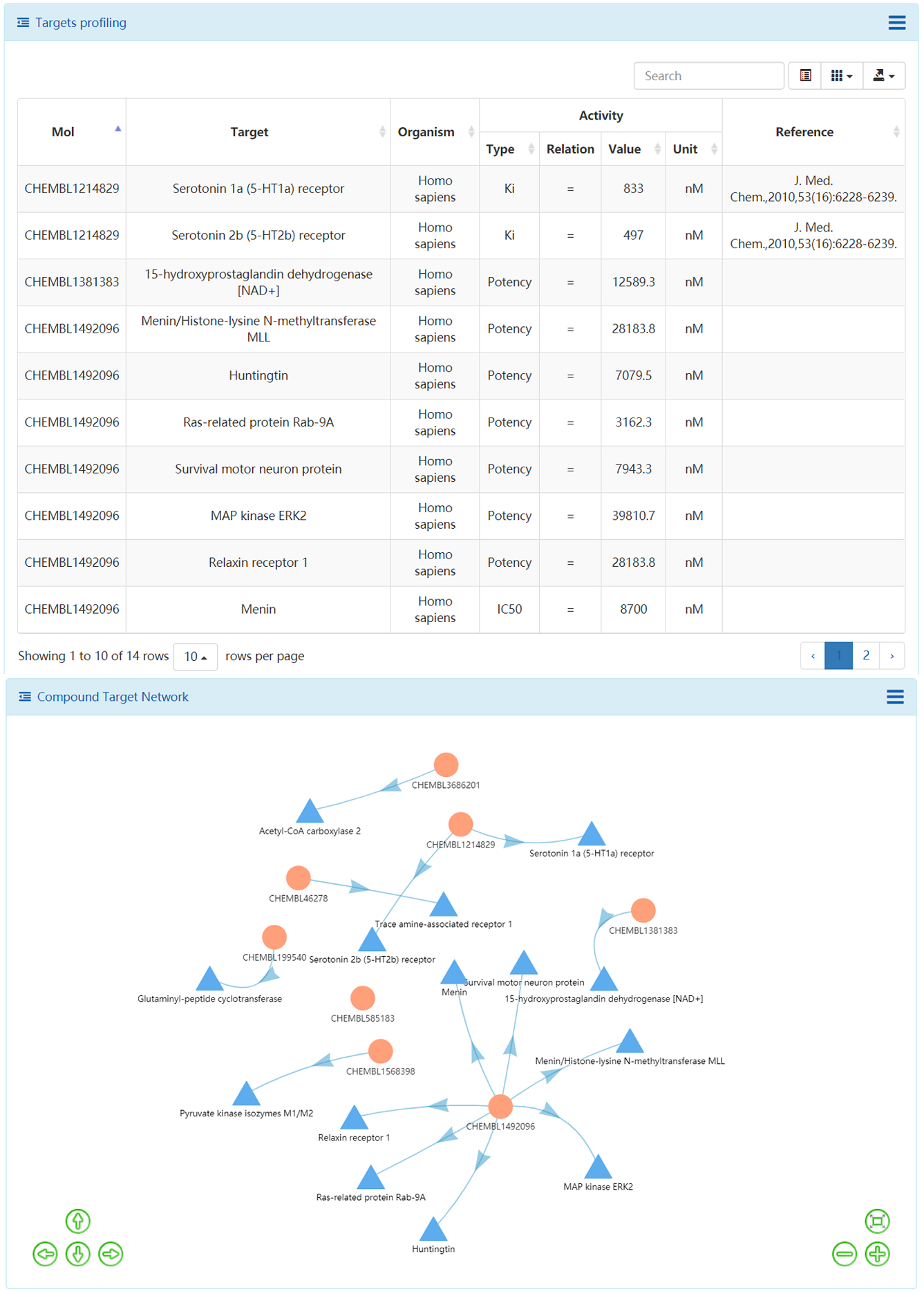
4.6 Target & Pathway
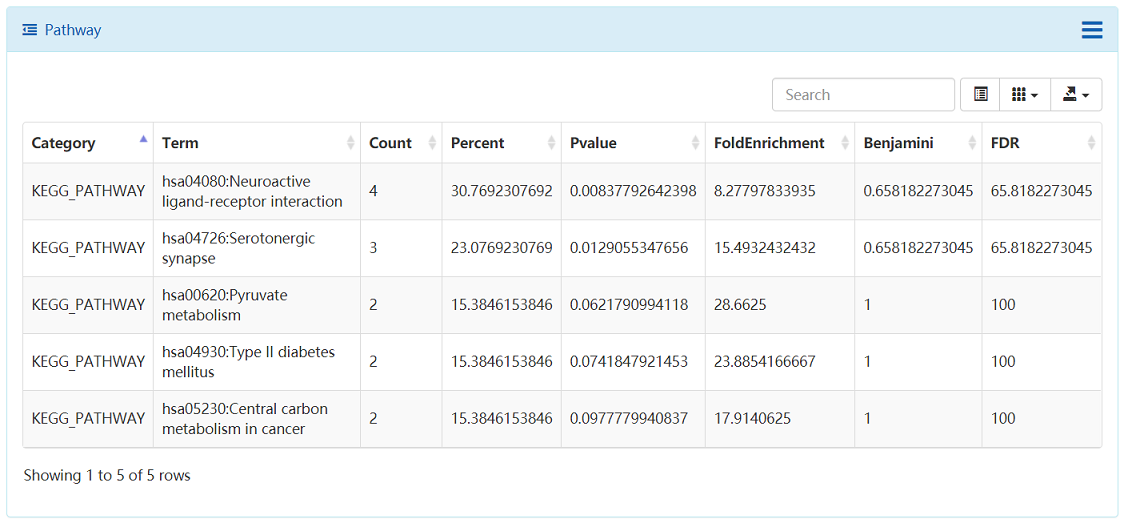
The ligands and bioactivity data in ChEMBL database were prepared and extracted. The Open Babel was used to generate the unique InChI Key for each structure, then using the InChI Key as query parameter to search against ChEMBL database, the bioactivity data such as Ki, Kd, IC50 and EC50, and corresponding references are extracted. All those results can be analyzed via a user-friendly table view. Those results are also can be downloaded for local analysis. The compound target relations were further illustrated using compound target network. The targets are further enriched to pathways and the KEGG pathways are summarized in table.
4. Databases and Tools
DenovoProfiling is constructed using various open databases and tools for different purposes. Here we appreciate their contributions and the tools names, the purposes and the links are list below.
| # | Tools | Purpose | Link |
|---|---|---|---|
| 1 | ChEMBL22 | target profiling | www.ebi.ac.uk/chembl |
| 2 | DrugBank | drug profiling | www.drugbank.ca |
| 3 | ChemDoodle Web | structure visulaization | web.chemdoodle.com |
| 4 | 3Dmol.js | molecular alignment results | 3dmol.csb.pitt.edu |
| 5 | D3.JS | descriptors distribution | d3js.org |
| 6 | Open Babel | chemical file format convertion | openbabel.org |
| 7 | PaDEL | properties and fingerprint calculation | padel.nus.edu.sg/software/padeldescriptor |
| 8 | MySQL | storage database | www.mysql.com |
| 9 | Golang | web server language | golang.org |
| 10 | R | calculate distance | www.r-project.org |
About us
Research Group for Gut Microbiome and Health (RGGMH) at Guangdong Institute of Microbiology was established by Dr. Liwei Xie and funded by grants from Guangdong Academy of Sciences and Guangdong Institute of Microbiology. The research group focuses on the AI-algorithm development and application in drug design, functional and health benefit small molecules and nature products design and screening, targeting gut microbial dysbiosis-associated metabolic diseases, such as Type 2 diabetes (T2D) and obesity.
Address
State Key Laboratory of Applied Microbiology Southern China,Guangdong Institute of Microbiology,
100 Xianlie Middle Rd, BLD58 2rd Floor,Guangzhou, Guangdong, China, 510070
Contact
Fax: +86-020-37656322
Telephone: +86-17688872852
Email: xielw@gdim.cn